11
THE WORLDWIDE PROTEIN DATA BANK
INTRODUCTION
The Protein Data Bank was established at Brookhaven National Laboratory (BNL) (Bernstein et al., 1977) in 1971 as an archive for biological macromolecular crystal structures. It represents one of the earliest community-driven molecular biology data collections. In the beginning, the archive held seven structures, and with each passing year a handful more were deposited. In the 1980s, the number of deposited structures began to increase dramatically. This was due to the improvements in technology for all aspects of the crystallographic process, the addition of structures determined by other methods, and changes in community views about data sharing. By the early 1990s, many journals required a PDB ID forpublication; now, virtually all journals require not only coordinates but also the primary experimental data be deposited with the PDB. As of December 2, 2008, the archive contained 54559 structures.
The initial goal of the PDB was to archive author-submitted structures determined by X-ray crystallography. Today, structures are deposited in the PDB that have been determined using X-ray crystallography, nuclear magnetic resonance (NMR), and, most recently, cryo-electron microscopy (cryoEM). In addition to the structural biologists who deposit data, the majority of PDB users are a diverse group of researchers in the biomedical sciences (biologists, chemists, physicists, etc.), as well as educators and students at all levels. To ensure that the PDB would remain a single uniform archive freely accessible via a collection of FTP tools, the worldwide PDB (wwPDB; www.wwpdb.org/) was formed in 2003 (Berman, Henrick, and Nakamura, 2003) with an agreement among three depositions centers in the United States, Europe, and Japan: the Research Collaboratory for Structural Bioinformatics (RCSB PDB), the Macromolecular Structure Database at the European Bioinformatics Institute (now called Protein Data Bank Europe (PDBe)), and the PDB Japan (PDBj). The BioMagResBank (BMRB), which archives biomolecular NMR data, joined the wwPDB in 2006. In this chapter, we describe the collection, validation, annotation, and distribution of data by the wwPDB. We additionally provide a brief description of the services offered by the member sites.
DATA ACQUISITION AND PROCESSING
Content of the Data Collected by the wwPDB
The PDB archive consists of entries containing three-dimensional Cartesian coordinates, information specific to the method of structure determination, and more recently experimental data. These include structure factors for X-ray experiments and constraints generated by NMR experiments. NMR depositions in the PDB contain links to the BMRB archive of web-accessible constraints, chemical shifts, and other data relevant to NMR structures. In the near future, volumes for electron microscopy will become a part of the PDB archive. Table 11.1 contains the general information that the wwPDB collects for all structures as well as the information specific to X-ray, NMR, and cryoEM experiments.
The definitions of the data items collected are in the PDB Exchange Dictionary (PDBx) (Westbrook et al., 2005a) that is based on the mmCIF syntax. The mmCIF dictionary contains 1,700 terms that define the macromolecular structure and the crystallographic experiment (Fitzgerald et al., 2005). PDBx contains additional terms for NMR and cryoEM. The BMRB-developed NMR-STAR Dictionary specifies additional data items linked to NMR structures. These terms were developed in collaboration with the depositors who are experts in these methods. Terms needed for tracking and other information management purposes are also contained in the PDBx.
Data Deposition Sites
Data are deposited to the PDB at one of the wwPDB member sites. Because it is critical that the final archive is kept uniform, the content and format of the final files, as well as the methods used to check them, are the same.
Each deposition to the PDB is represented by a PDBid—a four-character code. The PDBid is assigned arbitrarily and is an immutable reference to the structure. PDBids are never reused and remain the link between the structure and the literature reference that describes that structure. Experimental NMR data processed and archived at the BMRB are identified by a unique and immutable integer tag. Hyperlinks in the related PDB and BMRB files provide seamless access to all information.
The RCSB PDB (deposit.pdb.org/adit/) and PDBj (pdbdep.protein.osaka-u.ac.jp/adit/) use the program ADIT for data deposition and validation. PDBe processes data that are submitted to the site via AutoDep (autodep.ebi.ac.uk/) (Tagari et al., 2006).
BMRB sites at Madison (batfish.bmrb.wisc.edu/bmrb-adit/) and Osaka (nmradit. protein.osaka-u.ac.jp/bmrb-adit/) use ADIT-NMR to collect both coordinate and experimental data. The experimental data are processed by BMRB and the coordinate data by RCSB PDB.
After processing is complete, coordinates, structure factor files, and constraints from all the sites are sent to the RCSB PDB for inclusion in the archive.
Validation and Annotation
Validation refers to the procedure for assessing the quality of deposited atomic models (structure validation) and for assessing how well these models fit the experimental data (experimental validation). Annotation refers to the process of adding information resulting from the validation to the entry. The wwPDB validates structures and associated data by using accepted community standards.
TABLE 11.1. ContentofDatainthePDB
| Content of All Depositions |
| Source: specifications such as genus, species, strain, or variant of gene (cloned or synthetic); expression vector and host, or description of method of chemical synthesis |
| Sequence: Full sequence of all macromolecular components |
| Chemical structure of cofactors and prosthetic groups |
| Names of all components of the structure |
| Broad description of the function and/or the composition of the structure |
| Literature citations for the structure submitted |
| Three-dimensional coordinates |
| Keywords and experimental method |
| Additional Items for X-ray Structure Determinations |
| Temperature factors and occupancies assigned to each atom |
| Crystallization conditions, including pH, temperature, solvents, salts, and methods |
| Crystal data, including the unit cell dimensions and space group |
| Presence of noncrystallographic symmetry |
| Data collection information describing the methods used to collect the diffraction data including instrument, wavelength, temperature, and processing programs |
| Data collection statistics including data coverage, Rsym, I/sigma I, data above 1, 2, 3 sigma levels and resolution limits |
| Refinement information including R factor, resolution limits, number of reflections, method of refinement, sigma cutoff, geometry rmsd, sigma |
| Structure factors: H, k, l, Fobs, sigma Fobs, Intensity, sigma intensity, Flag for free R test |
| Additional Items for NMR Structure Determinations, |
| For an ensemble, the model number for each coordinate set that is deposited and an indication if one should be designated as a representative |
| Data collection information describing the types of methods used, instrumentation, magnetic field strength, console, probe head, and sample tube |
| Sample conditions, including solvent, macromolecule concentration ranges, concentration ranges of buffers, salts, antibacterial agents, other components, isotopic composition |
| Experimental conditions, including temperature, pH, pressure, and oxidation state of structure determination and estimates of uncertainties in these values |
| Noncovalent heterogeneity of sample, including self-aggregation, partial isotope exchange, conformational heterogeneity resulting in slow chemical exchange |
| Chemical heterogeneity of the sample (e.g., evidence for deamidation or minor covalent species) |
| A list of NMR experiments used to determine the structure including those used to determine resonance assignments, NOE/ROE data, dynamical data, scalar coupling constants, and those used to infer hydrogen bonds and bound ligands. The relationship of these experiments to the constraint files is given explicitly. |
| Constraint files used to derive the structure as described in the Task Force recommendations |
| Links (where available) to associated files at BMRB containing chemical shift, filtered constraint, and other NMR data |
| Additional Data Items for Cryoelectron Microscopy |
| Sample preparation, including aggregation state, concentration, buffer, pH, sample support, and description of vitrification procedure |
| 3D reconstruction procedure, including method, nominal and actual pixel size, resolution, CTF |
| correction method, magnification calibration, and text description of the reconstruction procedure 3D model fitting procedure, including fitting procedure type and program used, IDs of models used 3D model refinement, including whether refinement is done in real space or reciprocal space, type of |
| protocol, and refinement target criteria. If sample is a 2D or 3D array, symmetry and repeat parameters. If the entry contains a virus, details of the virus host including type, species, and growth cell, as well as details of the virus including type, isolate, and International Committee on Taxonomy of Viruses ID. |
In almost all cases, serious errors detected by these checks have been corrected through annotation and correspondence with the authors.
The wwPDB continuously reviews the validation methods used and will continue to integrate new procedures as they become available and are accepted as community standards.
Figure 11.1a shows the growth of PDB data, indicating how the complexity of structures released into the archive has increased over time.
Figure 11.1. Growth in the PDB archive as of December 2, 2008. (a) Growth chart of the PDB showing the total number of structures available in the PDB archive per year and highlighting example structures from different time periods. (b) Number of residues released in the PDB per year.
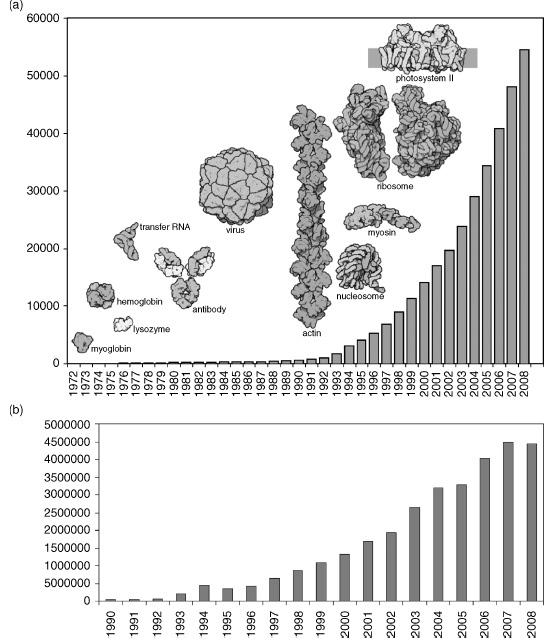
TABLE 11.2. Demographics of Data Released in the PDB (as of December 2, 2008)
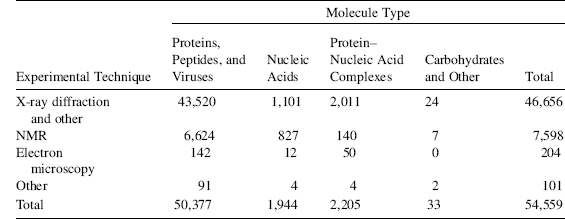
As of December 2, 2008, the PDB contained more than 54000 publicly accessible structures; of these entries, 46656 (86%) were determined by X-ray methods, 7598 (14%) were determined by NMR, and 204 were determined by cryoEM. Overall, 73% of the entries have released experimental data. As of this writing, in 2008 more than 99% of X-ray structures had structure factors deposited. For NMR structures, 98% were accompanied by restraint files and 44% by chemical shifts and other supporting data (Table 11.2).
Data Uniformity
A key goal of the wwPDB is to make the archive as consistent and error-free as possible. Before the files are released, all new depositions are reviewed by annotators. Errors found subsequent to release by authors and PDB users are addressed as rapidly as possible and the entries are revised.
The member sites of wwPDB have collaborated to remediate all the data in the PDB archive (Henrick et al., 2008; Lawson et al., 2008). In particular, the monomer components and the ligands have been examined and a new dictionary has been constructed that includes the stereochemistry, nomenclature, ideal coordinates, model coordinates as well as SMILES, and InChI representations. A Chemical Component Dictionary (www.wwpdb.org/ccd.html) contains the definitions for all components. All the instances of these components contained in the PDB entries have been matched against this dictionary and where necessary corrections have been made to the PDB files. The sequences, citations, and some experimental data items have also been corrected.
The corrected files are available at the wwPDB site (ftp://ftp.wwpdb.org). Coupled with this remediation effort has been a careful review of all annotation practices that are documented at (www.wwpdb.org/docs.html).
DATA ACCESS
FTP
The “PDB archive” is the collection of flat files that are maintained in three different formats: the legacy PDB file format (Bernstein et al., 1977); the PDB exchange format (PDBx) that follows the mmCIF syntax (Fitzgerald et al., 2005; Westbrook et al., 2005a) (mmcif.pdb.org); and the PDBML/XML format (Westbrook et al., 2005b) that is a direct translation of the PDB exchange format. Each wwPDB site distributes the same PDB archive via FTP. The archive is updated weekly.
Time-stamped snapshots of the PDB archive are added each year to ftp://snapshots. rcsb.org. They provide a frozen copy of the archive as it appeared at that time for research and historical purposes. Scripts are available to download all, or part, of a snapshot automatically.
Web Sites
In addition to providing access to the PDB archive, each wwPDB site provides services and resources that provide different views and analyses of the structural data contained within the PDB archive.
RCSB PDB
The RCSB PDB (www.pdb.org) (Berman et al., 2000) provides a web site with several functionalities. In addition to being able to download files in PDB, PDBx, and PDBML formats, there are different types of search capabilities. These include search by PDB ID, author, or keyword; search within a relational database that is populated by the data in the PDBx/PDBML files; or browsing characteristics that have been integrated from external resources (Deshpande et al., 2005). These include biological process, cellular component, and molecular function as defined by Gene Ontology (The Gene Ontology Consortium, 2000); enzyme classification, Medical Subject Headings (MeSH terms), source organism as defined by the NCBI Taxonomy, gene location from Entrez (Wheeler et al., 2006), and folds as classified by SCOP (Murzin et al., 1995) and CATH (Orengo et al., 1997). Many data items on the Structure Summary page for each structure are links to searches for other structures with similar properties. A variety of tabular reports can be made for groups of structures with information about the experiment, the chemistry and biology, and citations. Static pictures and interactive graphics are provided for each structure and protein-ligand interaction from the web site. Some of these interactive views use the molecular biology toolkit (MBT; mbt.sdsc.edu; Moreland et al., 2005). Avariety of summary statistics are available as histograms and in tabular form. Advanced searches can be performed on the structure data in combination with the data integrated from over 30 different external sources. Multimedia tutorials are available describing these features.
Also contained at the RCSB PDB site is the Molecule of the Month authored by David S. Goodsell. This feature describes a molecule in such a way that nonspecialists can learn about a particular molecular system. The BioSync (biosync.pdb.org) resource provides information about the characteristics of synchrotron beamlines (Kuller et al., 2002).
For application developers, a variety of Web Services are available to directly extract subsets of data.
PDBj
PDBj (www.pdbj.org) maintains a variety of services for querying, displaying, and analyzing PDB data. xPSSS, in addition to providing PDBid and keyword searches, also allows more sophisticated text-based searches. xPSSS takes advantage of PDBj’s native extensible markup language (XML) database by using XPath and XQuery to perform even very complicated queries efficiently. Structure Navigator can be used to locate PDB entries that are similar to a query structure or PDB ID. Structural similarity is defined by the newly developed scoreused in the stand-alone program ASHs (Alignment of Structural Homologues), which can be run or downloaded from www.pdbj.org/ASH/. ASH shows high sensitivity and selectivity in recognizing CATH or SCOP domains, without requiring large amounts of CPU (Standley, Toh, and Nakamura, 2004; Standley, Toh, and Nakamura, 2005; Standley, Toh, and Naka-mura, 2007). Sequence Navigator is a BLAST interface to PDBj. The input to Sequence Navigator can be a PDB ID or an amino acid sequence. Most of the query services described above can be executed as Web Services using the Simple Object Application Protocol (SOAP) or through web browsers. jV version 3 (www.pdbj.org/jV, formerly known as PDBjViewer) is a program to display molecular graphics of proteins and nucleic acids (Kinoshita and Naka-mura, 2004). The eProtS, Encyclopedia of Protein Structures, is an educational resource for learning biological functions and structural characteristics of protein molecules. The eProtS now contains explanations for a few hundred protein molecules that are of particular interest. eProtS is implemented as a Wiki so that motivated structural biologists can contribute articles for the proteins they have solved. Two derived databases are also available from PDBj. ProMode is a database of normal mode analyses (NMA) of proteins. eF-site (electrostatic surface of functional site) is a database for molecular surfaces of proteins’ functional sites, displaying the electrostatic potentials and hydrophobic properties together on the Connolly surfaces of the active sites, for analyses of the molecular recognition mechanisms. eF-seek is a service for searching the similar molecular surface of the known active sites (Standley et al.2008).
PDBe
PDBe (www.ebi.ac.uk/msd) (Velankar et al., 2005) has developed a variety of search and retrieval systems, some of which are summarized here. MSDChem (www.ebi.ac.uk/msd-srv/chempdb) and MSDsite (Golovin et al., 2004) (www.ebi.ac.uk/msd-srv/msdsite) extract the ligand binding information and characterize their chemical and geometrical environments using the MSD relational database thus allowing a user to obtain detailed information about a specific ligand or macromolecule-ligand interaction. MSDanalysis (www.ebi.ac.uk/ msd-as/MSDvalidate) allows statistical analysis of macromolecular structure. These include the following: MSDvalidate: the validation of structures within the PDB and uploaded files; MSDstatistics: the view of statistical information from the molecules in the PDB; MSDse-lection: the creation of selections of PDB molecules based on statistical distributions of molecular data; MSDresidue statistics: the analysis of the residue-based data from macro-molecular structure; MSDdatabase: a web interface that allows SQL queries to be made against the MSD database; and MSDmine: a fully featured analysis of the MSD database based on an SQL like interface.
PISA is an interactive tool for the exploration of macromolecular interfaces, prediction of probable quaternary structures (assemblies) and database searches of structurally similar interfaces and assemblies (Krissinel and Henrick, 2005). MSDMOTIF is a database that allows to perform fast Phi/Psi search, secondary structure patterns search, and small 3D structural motifs’ complexes search (www.ebi.ac.uk/msd-srv/msdmotif/). It also allows visualization as well as sequence multiple alignment and 3D multiple alignment.
MSDtemplate provides matching of a small number of amino acids by geometry to a protein (www.ebi.ac.uk/msd-srv/MSDtemplate). A template can be an active site, ligand environment, or any set of amino acids that are of interest. The MSD has a library of templates generated from data mining the PDB looking for statistically significant collections of residues. These templates from data mining include all the known active sites and metal binding sites.
BMRB
The BMRB (www.bmrb.wisc.edu) (Ulrich, Markley, and Kyogoku, 1989) archive can be searched by database ID, keywords, author, molecule name, sequence, sample conditions, or other characteristics. The website contains statistics and resources for the NMR community. BMRB archives data connected with an NMR structure as recommended by the wwPDB NMR Task Force for structural biology depositions: structural constraints, assigned chemical shifts, and NOESY spectra and/or peak lists. BMRB also supports more comprehensive NMR data depositions by members of structural genomics community, which may include additional primary (time domain) spectra and intermediate results.
The BMRB provides software and tools for validating NMR data and associated structures prior to deposition and tools for visualizing various types of NMR data with or without an associated three-dimensional structure. The BMRB is responsible for processing constraint files and for providing constraints to the PDB archive that are consistent with structural models and converted to machine-readable NMR-STAR format. Filtered restraints, further processed for use by test calculation protocols and longitudinal analyses and validations, are available online.
FUTURE
Structural biology is a fast evolving field that poses challenges to the collection, curation, and distribution of macromolecular structure data. Since 1998, the rate of deposition has tripled, and the total holdings have increased by a factor of five. The structures have increased in complexity with many molecular machines now part of the archive. In addition, the use of data harvesting has made it possible to collect more information about each structure. The challenge is to be able to continue to collect data rapidly and at the same time maintain quality.
The maintenance and further development of the PDB archive is a worldwide effort. The willingness of the global community to share ideas, software, and data provides a unique resource for biological research.
ACKNOWLEDGMENTS
The RCSB PDB is operated by Rutgers, the State University of New Jersey and the San Diego Supercomputer Center and the Skaggs School of Pharmacy and Pharmaceutical Sciences at the University of California, San Diego. It is supported by funds from the National Science Foundation, the National Institute of General Medical Sciences, the Office of Science, Department of Energy, the National Library of Medicine, the National Cancer Institute, the National Center for Research Resources, the National Institute of Biomedical Imaging and Bioengineering, the National Institute of Neurological Disorders and Stroke, and the National Institute of Diabetes and Digestive and Kidney Diseases.
EBI-MSD gratefully acknowledges the support of the Wellcome Trust
(GR062025MA), the EU (TEMBLOR, NMRQUAL, and IIMS), CCP4, the BBSRC, the MRC and EMBL.
PDBj is supported by grant-in-aid from the Institute for Bioinformatics Research and Development, Japan Science and Technology Agency (BIRD-JST), and the Ministry of Education, Culture, Sports, Science, and Technology (MEXT).
The BMRB is supported by NIH grant LM05799 from the National Library of Medicine.